米国食品医薬品局(FDA)による医療機器分類は、製品の市場投入可否を左右する最重要プロセスです。クラスI(低リスク)からクラスIII(高リスク)まで3段階に分かれるこの分類システムは、約1,700種類の機器タイプを16の専門分野に整理し、それぞれに適切な規制要件を課しています。しかし、その判定基準の複雑さゆえに、企業が誤った分類を採用してしまうケースが後を絶ちません。
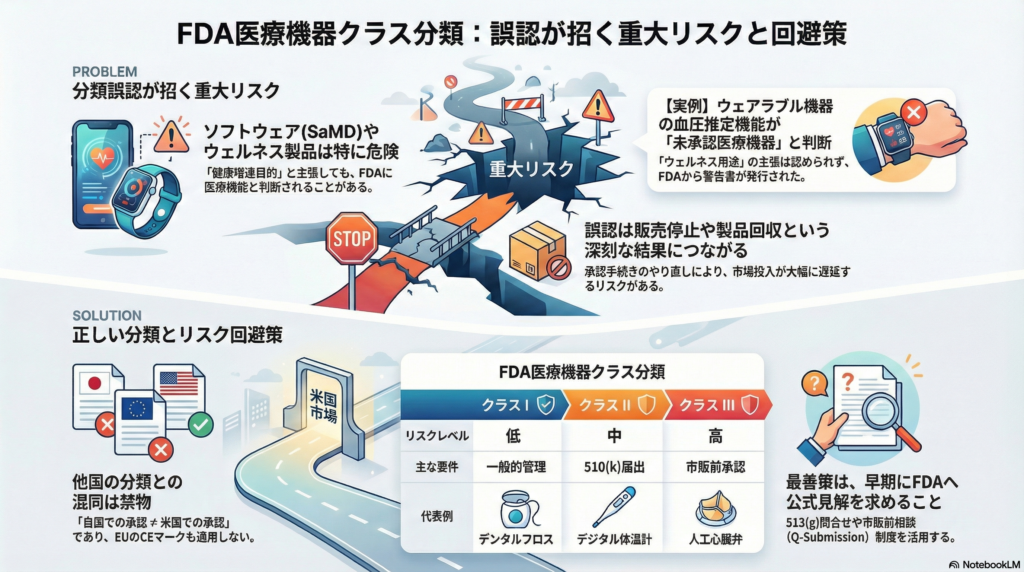
特にソフトウェア医療機器(SaMD)や境界領域製品では、「医療機器に該当するか否か」の線引き自体が曖昧になりがちです。2023年には心臓ポンプ遠隔監視ソフトウェアが「医療データシステムで規制対象外」と判断されていたにもかかわらず、FDA査察で未承認医療機器と認定され警告書が発出されました。2025年にはウェアラブル機器の血圧推定機能が「一般ウェルネス用途」として販売されたものの、FDAから医療機器機能と判断され同様の措置を受けています。
こうした分類誤認は、承認手続きのやり直し、製品回収、販売停止、さらには行政処分や罰金といった深刻な影響を企業にもたらします。本記事では、FDAクラス分類の構造と判定基準を整理したうえで、誤認が発生しやすいポイント、過去の実例、外国企業が特に注意すべき傾向まで包括的に解説します。
FDA医療機器分類の基本構造:クラスI・II・IIIとは
各クラスの定義と規制要件
FDAは医療機器をリスクレベルに応じて3つのクラスに分類し、それぞれ異なる規制要件を課しています。
**クラスI(低リスク機器)**は、患者に与えるリスクが最も低い機器群です。一般的管理(General Controls)と呼ばれる基本的な品質管理や表示義務のみで安全性が担保できるため、多くは市販前届出(510(k))が免除されています。代表例は歯間清掃用デンタルフロスです。
**クラスII(中等度リスク機器)**は、一般的管理に加えて特別管理(Special Controls)が適用される機器です。性能基準の順守や市販後監視などの追加要件があり、多くは510(k)届出によるFDA審査が必要となります。ただし一部のクラスII機器は510(k)免除の対象です。コンドームやデジタル体温計がこのクラスに該当します。
**クラスIII(高リスク機器)**は、生命維持や重大な医療行為に関わる最もリスクの高い機器です。FDAによる事前承認(PMA:Premarket Approval)が必要で、厳格な審査と臨床的証拠の提出が求められます。人工心臓弁や埋め込み型ペースメーカーがこのクラスに分類され、FDA承認なしには販売できません。
なお、1976年の医療機器規制導入時には情報不足により約25種の機器が一時的にクラスIIIに「仮置き」されました。法律上これらは36か月以内に再分類されるべきでしたが、実際には長年放置された事例も存在します。
分類を決定する2つの判定基準
FDA医療機器分類は、意図された使用目的(intended use)とリスクの程度という2つの軸で決定されます。
まず製品の用途・適応症に基づき、連邦規則集21 CFRの既存分類規則を照合します。該当する分類があればそのクラスに従います。この際、意図された使用目的における適応範囲(indications for use)が極めて重要です。たとえば「組織を切開するためのメス」と「眼科手術(角膜切開)のためのメス」では、後者のほうが高リスクと判断され分類が変わる可能性があります。
次に患者や使用者へのリスクの程度が評価されます。リスクが低ければクラスI、高ければクラスIIIに割り当てられる仕組みです。クラス分類によって市場投入前に必要な手続き(510(k)届出かPMA承認申請か)が決まり、各クラス共通の一般的管理は全機器に適用されます。
分類誤認が発生しやすい3つの境界領域
境界領域製品:医療機器か否かの判断が難しいグレーゾーン
医療機器の定義に当てはまるか否かが曖昧な「グレーゾーン製品」では、分類ミスが頻発します。
特に問題となるのがコンビネーション製品です。薬剤・生物製剤を含む医療機器では、主たる作用機序がどれかによって担当部門(CDRH:医療機器、CDER:医薬品、CBER:生物製剤)が決まります。この判断は容易ではなく、企業が独自に誤った区分で申請すると審査のやり直しを招きます。FDA内部にはコンビネーション製品課(Office of Combination Products)があり、企業は製品属性指定(Request for Designation, RFD)を提出することで主担当を明確化できます(60日以内に回答)。
また、製品が医療機器の定義に該当するか否か(主たる目的が人体の構造・機能への影響であり、主要作用が化学的作用によらないこと)も争点となります。近年ではFDAと企業の間で「これは医薬品ではなく機器か?」という解釈違いが訴訟に発展した事例も報告されています(Genus Medical社 vs FDA事件など)。
こうした境界領域では、類似製品の前例を調査し、早期にFDAと協議することが分類誤認を防ぐ鍵となります。
ソフトウェア医療機器(SaMD):法規上の例外と落とし穴
医療用途のソフトウェアは、FDA分類ルールの中でも特に誤認されやすい領域です。物理的ハードウェアを伴わないため一見すると医療機器らしくありませんが、「疾病の診断・治療や患者モニタリング」に用いられる機能を持てば医療機器に該当します。
ただし、ソフトウェアには法規上の例外や緩和措置も存在し、企業がこれを誤解するケースが多発しています。
一般的健康増進(General Wellness)用途の製品は、フィットネストラッカーや生活習慣改善アプリなど、リスクが低く医療目的を謳わない製品を指します。これらは医療機器から除外され得ますが、医療機能に踏み込むと対象外となります。
実例として、あるウェアラブルメーカーは血圧推定機能を「ウェルネス情報」として提供しましたが、FDAは「血圧測定それ自体が高血圧の診断に本質的に関連する」と判断し医療機器機能と認定しました。同社は疾病を明言しないマーケティングをしていたものの、FDAは「血圧の測定・推定は低血圧症や高血圧症の診断に関連するため一般的健康用途とはみなせない」と指摘し、当該機能をFDA未承認の医療機器機能と警告しました。
**医療データシステム(MDDS)や臨床意思決定支援(CDS)**も混乱の原因です。患者の医療データを記録・転送するだけのソフトや、医師の判断支援を目的とするソフトの一部は、条件を満たせば医療機器の定義から除外されます(21世紀治療法による規定)。しかしその適用基準は複雑です。
MDDSは「保存・表示・転送するのみ」の機能に限定され、CDS例外も医師が提示情報の根拠を独自に検証でき、最終判断を医師が下す場合に限られます。企業が自社ソフトは規制対象外と判断しても、実際には一部機能が基準を外れれば規制対象となります。
例として、心臓ポンプと連携する患者モニタリング用ソフトウェアを開発した企業が「これはCDS/MDDSで医療機器ではない」と主張したケースがあります。しかしFDA調査の結果、そのソフトは患者個別の心拍ポンプ異常を検知しアラーム通知する機能があり、生命に関わる状態を検出して医療介入を促すものと判断されました。このように「単なる表示・通知」か「能動的な診断支援」かの線引きは微妙で、企業の解釈ミスにより未承認のまま医療機器機能を提供してしまうリスクがあります。
AI搭載機器と新技術:規制の更新が追いつかない領域
人工知能(AI)や機械学習を用いた診断アルゴリズムなど、新しいデジタル技術も分類上の難所です。従来にない機能を持つため既存ルールに当てはめづらく、規制の更新が追いつかない領域ではグレーゾーンが生じやすいと指摘されています。
例えば、自己学習型の診断ソフトはリアルタイムで性能が変化するため、固定仕様を前提とした従来の分類体系ではリスク評価が難しくなります。規制当局もこうした製品に個別対応していますが、メーカー側の認識とFDA見解に齟齬が出やすい分野です。
この結果、将来的には最新技術をめぐる分類誤認が今まで以上に増える可能性が示唆されています。企業は専門知識を活用し、慎重な判断を下す必要があります。
FDAにおける分類決定プロセスと第三者の関与
FDA内部の分類プロセス:製品コードと分類パネル
医療機器のクラス分類は、FDA医療機器局(CDRH)が管轄し、法令で定められた分類規則に従って行われます。基本的には既存機器との同等性に基づき、製造企業が自社製品に該当する分類規則(21 CFR 862~892の該当条項)や製品コードを特定しFDAに届け出る形です。
FDAは約16の専門分野ごとに機器タイプを列挙した分類パネルと、それに対応する**製品コード(3文字コード)**を用意しています。各製品コードに該当クラスや規制要件が紐づけられており、企業はFDAのデータベースで製品名や用途から製品コードを検索し、対応するクラスと求められる手続きを確認します。
例えば「臨床用水銀体温計」は21 CFR 880.2920に分類規則がありクラスIIと規定され、製品コード「FLK」が割り当てられています。このように製品コード=分類であり、コードが決まれば自ずとクラスI~IIIが定まります。
もっとも、新規性が高く既存の分類規則に当てはまらない機器も存在します。その場合、基本的にはリスクが不明としてクラスIII相当(PMA要求)と見なされますが、メーカーは**デノボ(De Novo)**と呼ばれる申請を行い、新たにクラスIまたはIIの分類創設をFDAに求めることができます。FDAが承認すれば以後その機器タイプは低リスク機器として扱われます。
またメーカーが自社判断に迷う場合、513(g)問い合わせと呼ばれる正式照会制度でFDAに製品の分類・規制要件について見解を求めることもできます(回答にはユーザーフィーが必要)。さらに**市販前相談(Q-Submission)**制度を利用し、非公式にFDAと事前に協議して分類や必要な申請種別を確認することも推奨されています。
第三者認証機関の限定的な役割
米国における医療機器分類は基本的にFDAが一元的に決定します。欧州のような民間の「認定機関(ノーティファイドボディ)」が独自にクラス判定する制度はありません。したがって分類そのものを第三者が誤って認識するリスクは低いものの、第三者の関与が全く無いわけではありません。
FDAは一部のクラスII(低~中リスク)機器について、認定第三者機関によるプレマーケット審査を認めています。これは「510(k)第三者審査プログラム(Accredited Persons Program)」と呼ばれる制度で、FDAが認定した民間の審査機関がメーカー提出の510(k)申請を技術審査し、FDAに審査結果を勧告できる仕組みです。
この制度の目的は審査の迅速化ですが、重要な点は最終的な分類・承認判断権限はFDAに留保されていることです。第三者機関はあくまで技術レビューレポートをFDAに送付し、FDAが約30日以内に最終判断を下します。したがって、第三者機関が分類そのものを決めるわけではなく、分類判断や承認可否の最終責任は常にFDAにあります。
総じて、米国ではEUのような第三者認証による分類ミスという構図は少ないものの、企業自身やFDA審査官の解釈次第で分類が揺れるケースがあるため、製品の性質に照らした正確な分類選択が不可欠です。
過去に発生した分類誤認の実例と企業への影響
心臓ポンプ遠隔監視ソフトの誤認事例(2023年)
あるメーカーが提供した心臓ポンプ用リモート監視ソフトウェアは、自社では「医療データシステム(MDDS)や医師向け意思決定支援(CDS)ソフトに該当し、医療機器規制の対象外」と判断して販売されていました。
しかしFDA査察で調べたところ、そのソフトはポンプ装置から得た患者固有のデータを解析し、危険な心臓イベントを検知すると色分けされたアラームやメール通知で医療従事者に警告を発する機能を持つことが判明しました。FDAは「患者の生命に直結する状態を検知し時間的余裕のないアラームを出す機能は単なるデータ表示を超えた医療機器機能である」と判断し、このソフトはFDA未承認の医療機器そのものであると結論付けました。
その結果、2023年9月に当該企業に対し**警告書(Warning Letter)**が発出され、FDA承認(PMA)なしに販売した本製品は違法な無承認医療機器(FD&C法上の「adulterated」機器)に該当すると指摘されました。この警告書により企業は当該ソフトの出荷停止・是正措置を迫られ、新たにPMA申請を行うまで販売できない状況となりました。結果的に承認取得の遅延コストや信頼失墜のリスクを被り、社内の品質システム上の問題点も含めFDAから是正要求を受ける事態となっています。
ウェアラブル血圧モニタ機能の誤分類事例(2025年)
消費者向けフィットネスデバイスを製造するある米国企業は、自社のリストバンド型ウェアラブルに血圧推定機能を追加しました。この機能を「パフォーマンスや健康状態を理解するための新しい指標」と位置づけ、疾病の診断・治療を謳わない一般ウェルネス製品として売り出しました。
しかしFDAは、たとえ広告上明示的に疾病に言及していなくとも「血圧値を測定・表示する行為それ自体が高血圧・低血圧の診断に本質的に関連するため、一般的健康用途とは認められない」と判断しました。FDAは当該機能を医療機器(血圧計としての機能)と見なし、事前の510(k)届出またはPMA承認が必要であるにもかかわらずそれらを怠って市販したとして、2025年7月に当該企業(WHOOP社)に警告書を発行しました。
警告書では、この製品が適切な事前許可なく市販されたためFederal Food, Drug, and Cosmetic Act違反となる「adulterated(無承認流通)」かつ「misbranded(不適正表示)」デバイスに当たると指摘されています。この措置により企業は問題の血圧機能の提供を中止せざるを得ず、今後FDAと係争になる可能性も報じられています。
この事例は、一般向けヘルスケア製品であっても医療目的と見做されうるラインを誤ると迅速にFDAの介入を招くこと、そして米国市場ではマーケティング上の表現よりも製品機能そのものが厳しく精査されることを示しています。企業にとっては追加の規制対応コストや機能撤回による市場機会損失、ブランドイメージへのダメージといった負の影響が現実のものとなりました。
経頭蓋電気刺激(CES)機器の長期戦事例(1990年代~2010年代)
米国企業Alpha-Stim社の扱う経頭蓋電気刺激(CES)デバイスのケースは、分類誤認が企業と業界に長年影響した例として特筆されます。CES機器は不安・抑うつ・不眠症などの治療補助として微弱電流を流す装置ですが、FDAは1976年の規制開始時に十分な情報が無いとしてこれを一時的にクラスIIIに分類しました。
本来低リスクで安全な機器にもかかわらず厳格なPMA承認が要求されたため、市場では「CESはペースメーカー並みに危険なデバイスなのか?」という誤解が生じ、医療従事者や保険会社、患者の間に不信感が広まりました。Alpha-Stim社はこの誤った高リスク分類の是正を求めて1990年代からFDAと争いましたが、再分類請願や治験データ提出を繰り返しても審査は遅々として進まず、実に22年間も解決しませんでした。
この間、同社はクラスIIIの規制負担に苦しみ、業界全体も「ブラックボックス警告」を背負ったような状態で停滞を余儀なくされました。資金力の乏しい中小企業にとってフルPMA要求は重荷で、Alpha-Stim社は存亡の危機に瀕したといいます(当時は別用途の痛み治療向けクラスII適応で何とか収益を維持)。
政府説明責任局(GAO)も2009年に報告書で、FDAが1976年以来「一時的クラスIII」とした機器の多くを期限内に再分類せず放置した問題を指摘しました。最終的に2014年になってFDAはCESをクラスIIIから引き下げ再分類する意向を表明し(提案規則の公表)、長年の係争はようやく終止符に向かいました。
しかしこの間に費やした法務費用・試験コストは莫大で、同社はビジネスチャンスや米国内市場シェアを失いかけました。また規制当局への不信感から業界全体の士気も下がり、「FDAの定義や基準の適用が一貫性を欠き恣意的だった」と批判されています。この事例は分類ミスが企業に与える長期的影響(コスト、遅延、レピュテーション悪化)を物語っています。
分類誤認がもたらす経営リスク
上記のように、分類の誤認は企業にとって製品戦略の根幹を揺るがす事態となり得ます。FDAから警告書を受けた場合、速やかな是正措置を講じなければさらなる措置(罰金や差し止め命令、製品差押え等)につながることがあります。また自主的に市場から製品を回収しなければならないケースや、再申請まで販売停止となるケースもあります。
無承認販売は重大な法令違反と見なされるため、意図的な場合には民事罰のみならず刑事罰の対象ともなり得ます。実際に近年、FDAと司法省は無承認医療機器販売に関与した企業に数百万ドル規模の制裁金を科した例も報告されています(警告を無視し販売継続したコンタクトレンズ製造業者への罰金など)。
このように分類の誤りは企業に計り知れないコストとリスクをもたらすため、事前に適切な分類を行い、必要なFDA手続きを踏むことが何より重要です。
外国企業(日本企業を含む)が陥りやすい分類ミスの傾向
他国の分類との混同
各国・地域で医療機器のクラス分類基準は異なります。例えば欧州連合(EU)ではリスクに応じてクラスI(低リスク)からクラスIII(高リスク)+IVDと細分化されていますが、EUクラスとFDAクラスは名称こそ似ていても対応関係は単純に一致しません。
にもかかわらず、「EUでクラスIIaだからFDAでも中リスク程度だろう」と安易に対応付けしてしまい、FDAでは実はクラスIII相当の厳格管理が必要だった、といった誤りが起こります。特にFDAは「意図された用途(intended use)」のわずかな違いで分類が変わるため、外国企業が自社製品の用途を十分に精査せずに近似のFDA分類に当てはめるとミスを犯しがちです。
EUのような包括的ルール適用とは異なり、FDAは個別の製品コード・適応で分類するため、外国企業はこの点を理解しておく必要があります。
承認・届出要否の誤解
海外で承認・認証を得ている製品について、「自国で認められたから米国でも書類提出なしで売れる」と誤解するケースがあります。典型的なのはCEマーキング取得製品を米国に持ち込む際、「CE取得=安全と認められているから510(k)は不要だろう」という思い込みです。
しかしFDAは他国認証をそのまま認めることはなく、米国独自の分類評価と申請を要求します。CEマーク品でも米国では510(k)届出が必要な場合が多々ありますし、逆にEUでクラスI自己宣言だったものが米国では製品コード上510(k)必要なクラスIIだったという例もあります。
このように各国規制をマッピングしようとすると齟齬が生じるため、外国企業は市場ごとに分類・手続きを再確認することが不可欠です。
FDAの定義やガイダンスへの不慣れ
日本を含む海外企業は、FDA特有の概念や用語に不慣れなことからミスをしやすい傾向があります。例えば「medical device(医療機器)の法的定義」や「intended use(意図された使用目的)」の捉え方が自国と異なり、機器に該当するかどうかの認識を誤るケースがあります。
日本企業の場合、医薬品医療機器等法下での機器分類や承認制度に精通していても、FDAの510(k)/PMA区分や513(g)制度など初めて直面する手続きに戸惑うことがあります。また言語や書類作成の違いから、FDAへの提出資料で適応症の表現が不適切となり意図せぬ高リスクと見なされるリスクもあります。
実際、日本の規制当局(PMDA)では「申請書の小さな誤記や不備もその企業の信頼性問題と受け取り、内容を厳しく精査する」という文化があり、これは裏返せば日本企業が海外当局向け申請で軽微なミスを犯すと過度に警戒される可能性も示唆しています。
外国企業が取るべき対策
以上の傾向から、外国企業には早い段階でFDA規制専門家に相談し、分類や申請戦略を練ることが推奨されます。例えば製品が医療機器に該当するか微妙な場合は早めに513(g)問い合わせやプレサブミッションを行い、FDA見解を確認することで誤った路線で開発・販売計画を進めるリスクを減らせます。
また現地の法律用語やガイダンス文書を正確に解釈できる人材を確保し、「自国ではOKだったから大丈夫」の思い込みを避けることが重要です。グローバル企業は各市場のルール差異に精通したチームを置き、例えばEU vs USのクラス対応表を鵜呑みにせず個別にCFR分類を確認する、申請資料中の表現が意図せぬ医療効果を示唆していないかチェックする、といった細心の注意が求められます。
これらを怠ると、せっかく他国で軌道に乗った製品が米国で予期せぬ発売遅延や追加試験要求に直面し、ビジネス上大きな機会損失を被りかねません。特に米国は市場規模が大きい反面、規制遵守に厳格なため、外国企業ほど謙虚に専門知識を取り入れて万全の準備をすることが成功の鍵と言えます。
まとめ
FDA医療機器分類は、製品のリスクレベルに応じてクラスI(低リスク)からクラスIII(高リスク)まで3段階に区分され、それぞれ異なる規制要件が課されます。分類は「意図された使用目的」と「リスクの程度」という2つの判定基準に基づいて決定されますが、その複雑さゆえに企業が誤った分類を採用するケースが後を絶ちません。
特に境界領域製品、ソフトウェア医療機器(SaMD)、AI搭載機器など新技術領域では、「医療機器に該当するか否か」の線引き自体が曖昧になりがちです。過去には心臓ポンプ遠隔監視ソフトやウェアラブル血圧モニタ機能が規制対象外と誤認され、FDAから警告書を受けた事例が発生しています。また経頭蓋電気刺激(CES)機器のように、誤った分類が22年間も放置され企業に深刻な影響を与えた例も存在します。
分類誤認は、承認手続きのやり直し、製品回収、販売停止、行政処分、罰金といった深刻な影響を企業にもたらします。外国企業は他国の分類との混同、承認・届出要否の誤解、FDAの定義やガイダンスへの不慣れといった要因から特にミスを犯しやすい傾向があります。
これらのリスクを回避するには、早い段階でFDA規制専門家に相談し、513(g)問い合わせやプレサブミッションを活用してFDA見解を確認することが不可欠です。「自国ではOKだったから大丈夫」の思い込みを避け、市場ごとに分類・手続きを再確認する姿勢が、米国市場での成功の鍵となります。
コメント