はじめに:FDA警告書が示すサプリメント業界のリスク
米国FDAが発出する警告書(Warning Letter)は、サプリメント業界にとって最も重要な規制動向の指標です。2025年10月下旬から2026年1月中旬までの約3か月間に発出された警告書を分析すると、業界全体で繰り返される典型的な違反パターンが浮かび上がります。
本記事では、直近の警告事例から抽出した主要な違反カテゴリと、実務者が今すぐ取り組むべき対策について詳しく解説します。FDA規制への理解を深め、自社のコンプライアンス体制を見直すための実践的なガイドとしてご活用ください。
直近90日間の警告書:3つの重大違反事例
ケース1:衛生管理の失敗がもたらした全製品回収
2025年11月13日、ケンタッキー州のPan-African Food Distributors Inc.とEast Africa Boutique LLCに対して警告書が発行されました。この事例では、サプリメントを含む食品の保管倉庫において、ネズミや虫の大量発生という深刻な衛生不良が発覚しています。
連邦食品医薬品化粧品法(FD&C Act)402(a)(4)条に基づき、製品は「不衛生な環境で保管された不良品(adulterated)」と判断されました。州保健当局による操業停止命令とFDAの警告により、施設内の全製品が回収・廃棄される事態に発展しています。
この事例が示すのは、衛生管理の失敗が事業継続そのものを脅かすリスクです。倉庫や製造施設の衛生状態は、日常的なモニタリングと記録管理が不可欠であることを改めて浮き彫りにしました。
ケース2:品質管理体制の根本的欠如
2025年12月23日、ミシガン州のLXR Biotech, LLCに対する警告書では、エナジー系サプリメント飲料の製造所における複数のGMP違反が指摘されました。特に重大だったのは、21 CFR Part 111(サプリメントGMP規則)の基本要件を満たしていなかった点です。
具体的には、製品ごとの品質規格(成分の同一性・純度・強度・組成)を設定していない、原料ごとの受け入れ試験規格を定めていない、科学的に妥当な試験方法を採用していないといった、品質管理の根幹に関わる不備が列挙されています。
これらの違反により、製品はFD&C Act 402(g)(1)条のGMP要件不遵守による不良品とみなされました。この事例は、GMP遵守が単なる書類作業ではなく、製品の安全性と品質を担保する実質的なシステムであることを示しています。
ケース3:臨床試験における規制上の認識不足
2026年1月8日、カリフォルニア州のProdrome Sciences USA, LLCに発出された警告書は、サプリメントと医薬品の境界線に関する重要な教訓を含んでいます。
同社はオメガ3系サプリメント「ProdromeNeuro™ Oil」を用いて、アルツハイマー関連の認知低下に対する臨床試験を実施しました。しかし、必要な治験申請(IND: Investigational New Drug)を提出していなかったことが問題視されています。
疾病の治療・予防を目的として使用される製品は、法律上「医薬品」と見なされます。たとえサプリメントとして販売する意図がなくても、研究目的での使用であっても、疾病への効果を検証する臨床試験を行えば、その製品はFDA未承認新薬として扱われる可能性があります。
この事例は、研究段階においても規制要件を正確に理解する必要性を示しています。
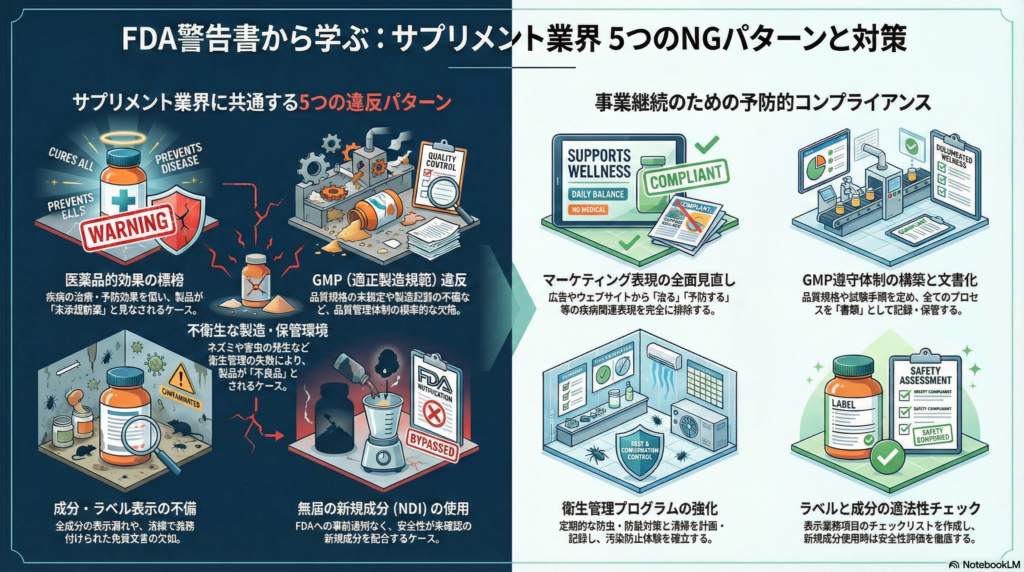
サプリメント業界に共通する5つの違反パターン
パターン1:未承認医薬品としての宣伝・販売
最も頻繁に見られる違反は、サプリメントに疾病の治療・予防効果を謳う行為です。「心血管疾患を治療できる」「糖尿病の症状を改善する」といった表現を使用すると、その製品は法律上、未承認新薬(unapproved new drug)と見なされます。
サプリメントとして許容されるのは、構造機能表示(身体の正常な構造や機能をサポートする表現)のみです。疾病への効果を示唆する表現は、たとえ間接的であっても規制違反となる可能性があります。
マーケティング資料、ウェブサイト、ソーシャルメディア投稿など、あらゆるチャネルでの表現が審査対象となるため、包括的なレビュー体制が必要です。
パターン2:GMP違反(製造・品質管理の不備)
21 CFR Part 111で定められたサプリメント製造管理基準への違反も、警告書の主要な発出理由です。特に以下の要素が欠如している事例が目立ちます。
製造記録の不備または欠如、品質試験の実施体制の不在、製品規格の未設定または不適切な設定、原料受け入れ試験の省略、試験方法の科学的妥当性検証の欠如などが典型的な違反内容です。
これらの違反により、製品は402(g)(1)条に基づき「GMP不遵守による不良品」と判断されます。中小企業では品質管理部門の独立性確保が難しい場合がありますが、規模に関わらずGMP遵守は必須要件です。
パターン3:不衛生な製造・保管環境
衛生管理の失敗は、最も深刻な結果をもたらす違反の一つです。ネズミの侵入、害虫の混入、設備の汚染などが発見されると、製品は402(a)(4)条により「不衛生による不良品」とされます。
実際の警告事例では、倉庫内に広範なネズミと虫の存在が確認され、全製品の回収に至ったケースがあります。このような状況は、防虫・防鼠対策の不備、清掃記録管理の欠如、定期的なモニタリング体制の不在などから生じます。
FDA査察で不衛生と判断されれば、即座の操業停止や製品回収に発展する可能性があります。日常的な衛生管理と記録保持が、事業継続の基盤となります。
パターン4:成分および表示ラベルの不備
製品ラベルに必要情報が欠落している、または誤表示がある場合も違反となります。典型的な不備には以下のようなものがあります。
全成分の正確な表示の欠如、用法用量や警告文の不記載、構造機能表示に必要な免責文言(DSHEAディスクレーマー)の欠如などです。
構造機能表示を行う場合、「これらの表現はFDAによる評価を受けておらず、本製品は疾病の診断・治療・予防を目的としない」旨の免責文言が必須です。現在FDAは免責文言の全パネル記載義務の緩和を検討中ですが、現行規則では該当箇所ごとに明記することが求められています。
ラベル表示の適法性は、製品発売前の最終チェック項目として徹底的に確認する必要があります。
パターン5:無届の新規成分の使用
市場実績や安全性情報が不十分な新規成分(NDI: New Dietary Ingredient)を含む製品も違反対象です。1994年以前に米国市場で使用実績のない成分をサプリメントに配合する場合、FDAへの事前通知が必要です。
この手続きを経ずに販売すると、402(f)(1)(B)条により「安全性証明の不備による不良品」とみなされます。最近では、ベニテングタケ(毒性を持つキノコ)由来成分を含む製品が、この規定により違法なサプリメントと判定され、FDAから警告を受けた事例があります。
新規性のある素材、特にキノコ由来成分やハーブ類を使用する際は、その成分の使用履歴を慎重に調査し、必要に応じてNDI通知を行うことが重要です。
実務者が今すぐ取り組むべき7つの対策
対策1:マーケティング表現の全面見直し
製品説明、広告、ウェブサイト、ソーシャルメディアなど、あらゆるチャネルでの表現を体系的にレビューします。「◯◯病に効く」「症状を治す」「予防する」といった疾病効果を示唆する文言は即座に削除または修正が必要です。
健康強調表示は構造機能に限定し、「免疫システムをサポート」「健康的なエネルギーレベルの維持」など、身体の正常な機能に言及する表現に留めます。
法務チームまたは規制専門家による定期的なレビュー体制を確立し、新しいマーケティング素材はすべて発表前に審査を受けるプロセスを導入してください。
対策2:GMP遵守体制の構築と文書化
21 CFR Part 111の各要件を満たす体制を構築します。特に重要なのは以下の要素です。
製品ごとの品質規格(成分の同一性・純度・強度・組成)の明確な設定、原料受け入れ時の試験規格と手順の確立、科学的に妥当な試験方法の採用と検証、製造記録の詳細な作成と保管、品質管理部門の独立性確保などが必須です。
これらの活動は単に実施するだけでなく、「書類による証拠」として残すことが重要です。FDA査察では、口頭での説明よりも文書化された記録が重視されます。
対策3:衛生管理プログラムの強化
製造所と倉庫の衛生環境を維持するための包括的なプログラムを実施します。具体的には、防虫・防鼠対策の定期的な実施と効果検証、清掃スケジュールの策定と記録管理、設備・器具の洗浄手順の標準化、汚染の兆候(虫の死骸、齧歯動物の排泄物等)に対する即時対応プロトコルの確立などが含まれます。
万一汚染の兆候が見つかった場合は、出荷停止、徹底洗浄・消毒、根本原因分析、再発防止策の実施という一連の対応を迅速に行う体制を整えておくことが重要です。
対策4:ラベル表示の適法性チェックリスト作成
製品ラベルに含めるべき情報をチェックリスト化し、発売前の最終確認に使用します。チェック項目には、全含有成分の正確な表示(一般名と学名)、成分量の適切な単位表示、用法・用量の明確な記載、保存方法と賞味期限、必要な警告文(アレルゲン情報など)、構造機能表示を行う場合のDSHEA免責文言などが含まれます。
免責文言の表示場所については、現時点では該当箇所ごとに表示することが求められていますが、FDA規制の動向を継続的にモニタリングし、変更があれば速やかに対応できる体制を整えてください。
対策5:新規成分使用時の安全性評価手順
新規性のある素材を製品に使用する前に、以下の手順を踏みます。
当該成分が米国市場で1994年10月15日以前から使用されていたかの文献調査、使用実績がない場合はNDI通知の必要性評価、安全性データの収集と評価(毒性試験、臨床データなど)、必要に応じた専門家への相談などです。
特にキノコ由来成分、エキゾチックなハーブ、新規の植物由来成分などは、慎重な安全性評価が必要です。安全性に合理的な確証がないまま販売すれば、違法な成分混入とみなされるリスクがあります。
対策6:臨床研究実施時の規制要件確認
サプリメントの効果を検証する臨床研究を計画する場合、その研究デザインと目的が規制上どう解釈されるかを事前に評価します。
疾病の改善、治療、予防を主要評価項目とする研究は、そのサプリメントを法的に「医薬品」として扱う可能性があります。このような研究を実施する場合は、治験薬(IND)申請、IRB(治験審査委員会)承認など、医薬品相当の手続きが必要になる場合があります。
研究段階であってもFDA規制の適用を受ける可能性があるため、臨床研究開始前に規制専門家や法務チームに相談することを推奨します。
対策7:継続的な規制動向のモニタリング体制
FDAの警告書、ガイダンス文書、規制変更の提案などを定期的に確認する体制を確立します。具体的には、FDA Warning Letters Database(食品・サプリメント分野)の月次レビュー、FDA Constituent Updatesやプレスリリースの購読、業界団体や専門家によるセミナー・ウェビナーへの参加、規制変更に対する社内対応手順の策定などが有効です。
規制環境は常に変化しており、最新の情報を把握することがコンプライアンス維持の基礎となります。
まとめ:予防的コンプライアンスが事業継続の鍵
直近90日間のFDA警告書分析から明らかになったのは、サプリメント業界における違反パターンには明確な傾向があるということです。医薬品的表現の使用、GMP体制の不備、衛生管理の失敗、ラベル表示の不適切さ、新規成分の無届使用という5つの主要パターンを理解し、実務レベルで対策を講じることが重要です。
警告書を受け取ってから対応するのではなく、予防的なコンプライアンス体制を構築することが、事業の持続的成長と信頼性確保につながります。本記事で紹介した7つの対策を参考に、自社の体制を見直し、継続的な改善に取り組んでください。
FDA規制への適切な対応は、単なるリスク回避ではなく、消費者の安全と信頼を守る企業責任の実践でもあります。規制遵守を競争優位性の源泉と捉え、業界全体の質の向上に貢献していきましょう。
コメント