はじめに:医療機器サイバーセキュリティが法的義務になった背景
医療機器のネットワーク接続が一般化する中、サイバー攻撃による患者安全へのリスクは現実的な脅威となっています。米国FDA(食品医薬品局)はこれまでガイダンスによる推奨事項でセキュリティ対策を促してきましたが、2023年3月29日、状況は大きく変わりました。包括歳出法2023のSection 3305により連邦食品医薬品化粧品法にSection 524Bが追加され、サイバーセキュリティ対策が法的要件として義務化されたのです。
本記事では、この規制強化が医療機器メーカーの開発プロセスにどのような影響を与えているのか、実務対応のポイントを含めて解説します。
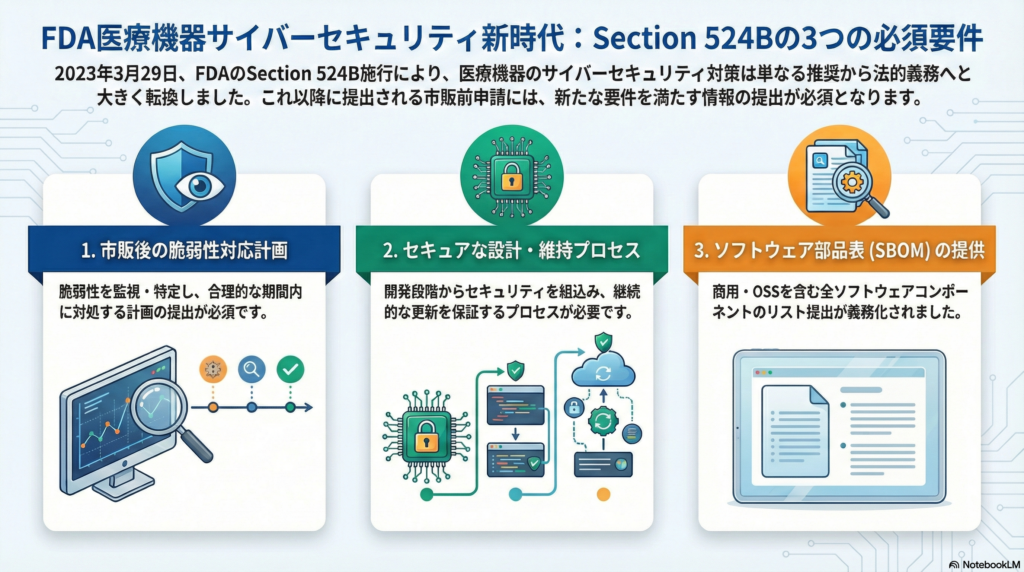
Section 524Bが定める三つの必須要件
Cyber Deviceの定義と適用範囲
Section 524Bでは、ソフトウェアを含みインターネット接続が可能で、サイバー脅威に脆弱となり得る技術的特徴を持つ機器を「Cyber Device」と定義しています。現代のネットワーク対応医療機器の大半がこの定義に該当するため、適用範囲は広範です。
2023年3月29日以降に提出される市販前申請(510(k)、PMA、De Novo等)には、以下三つのサイバーセキュリティ情報の提出が法的に義務付けられています。
1. ポストマーケット脆弱性対応計画
製品の市場投入後におけるサイバー脆弱性やエクスプロイトを監視・発見し、適切に対処する計画の提出が必要です。特に重要なのが協調的脆弱性開示(CVD: Coordinated Vulnerability Disclosure)の仕組みです。
外部の研究者やユーザから脆弱性報告を受け付ける窓口の設置、報告を受けた後の社内評価プロセス、そして合理的な期間内での対応策実施までの一連の手順を文書化することが求められます。これにより、セキュリティ研究者との協力関係を構築し、脆弱性の早期発見と対処が可能になります。
2. セキュア設計・維持のプロセス
機器および関連システムがサイバーセキュアであることを合理的に保証するためのプロセスと手順の確立が必要です。具体的には、製品ライフサイクルを通じてセキュリティを確保し、市販後にアップデートやパッチを提供可能にする設計が求められます。
このプロセスには、脅威モデリング、リスク評価、セキュリティアーキテクチャ設計、実装、テスト、そして継続的な改善までが含まれます。設計段階からセキュリティを組み込む「Security by Design」の考え方が、単なるベストプラクティスから法的要件へと格上げされた形です。
3. SBOM(ソフトウェア部品表)の提供
デバイスに含まれるすべてのソフトウェアコンポーネントのリスト提出が義務化されました。商用ソフトウェア、オープンソースソフトウェア(OSS)、オフザシェルフ部品を含む全構成要素について、名前、バージョン、ベンダー等の情報を記載したSBOMの提出が必要です。
SBOMにより、各ソフトウェア部品に既知の脆弱性が報告された際、製品への影響を迅速に評価できるようになります。特にOSSやサードパーティ製ソフトウェアは頻繁に脆弱性が公表されるため、SBOMの管理は継続的なセキュリティ維持に不可欠です。
プレマーケットガイダンスの大幅更新
2023年版から2025年版への進化
FDAは2023年9月27日に最終ガイダンス「Cybersecurity in Medical Devices: Quality System Considerations and Content of Premarket Submissions」を公開し、2014年の旧ガイダンスを置き換えました。さらに2025年6月27日にはSection 524Bへの言及を追加した改訂版が発行され、法的要件との整合性が明確化されています。
品質システムへのセキュリティ統合
ガイダンスの核心は、サイバーセキュリティを品質マネジメントシステム(QMS)や設計開発プロセスに統合する重要性です。Secure Product Development(SPD)フレームワークの活用により、製品開発ライフサイクル全体でセキュリティリスクに対処することが推奨されています。
リスクマネジメント、セキュリティアーキテクチャ設計、サイバーセキュリティテスト(脆弱性スキャン、ペネトレーションテスト、ファジング等)を開発プロセスに組み込み、設計段階からセキュリティを確保するアプローチが求められます。
リスクベース評価の明確化
産業界からのコメントを受け、最終ガイダンスにはサイバーセキュリティリスク評価に関する新セクションが追加されました。リスクのスコアリング方法、対策前後のリスク評価、受容基準など、リスクベースで対策優先度を決定する具体的な考え方が示されています。
これにより、限られたリソースの中で最も重要なセキュリティ対策に集中でき、効率的なリスク低減が可能になります。
提出すべき申請書類の具体的内容
サイバーセキュリティ計画書・リスクマネジメント文書
製品のセキュリティ目標や設計段階の脅威モデル分析、リスク評価結果、リスク低減策の一覧などを含む包括的な文書が必要です。脅威モデルでは、デバイスの使用シナリオや接続環境を分析し、想定される攻撃ベクトル(不正アクセス、データ改ざん、サービス妨害など)を洗い出します。
セキュリティアーキテクチャ設計図
デバイスのネットワーク構成図やデータフロー図、セキュリティ制御の実装ポイントを示す資料が求められます。アクセス制御、認証メカニズム、暗号化の実装箇所など、具体的なセキュリティ対策の設計を視覚的に示すことで、審査官が製品のセキュリティ態勢を理解しやすくなります。
テスト計画と結果
実施したサイバーセキュリティ試験の概要と結果、残存リスクの説明が必要です。脆弱性スキャンや侵入テストの手法、発見された脆弱性とその対処方法、対処しきれなかった残存リスクとその正当化理由などを文書化します。
アップデート管理計画
将来のソフトウェアアップデートやパッチ配信方法、頻度、重要度に応じた対応策の説明が求められます。重大な脆弱性発見時には速やかな緊急パッチ配布を可能にする仕組み、定期的なセキュリティ更新のスケジュール、アップデートの安全性を担保する方法(電子署名検証など)を明示する必要があります。
ユーザ向けセキュリティ情報
医療機関やエンドユーザに提供するデバイスのセキュリティに関する留意事項も必須です。ソフトウェア更新手順、推奨ネットワーク設定、サイバーリスクに関する警告文などを含むラベリング情報を準備します。
SBOM管理の実務対応
SBOM自動生成ツールの導入
手作業でのSBOM作成・更新は非現実的なため、開発環境にSBOM自動生成ツールを組み込む動きが広がっています。ソースコード管理システムと連動して依存ライブラリを解析しSBOMを出力するソフトウェアや、生成されたSBOMを用いて継続的に脆弱性スキャンを行うサービスの利用が進んでいます。
標準フォーマットの活用
業界ではSBOMの標準フォーマット(SPDX、CycloneDX等)の採用が進んでいます。標準化により、サプライヤからのSBOM提供や、複数製品にまたがるSBOM統合管理が容易になります。NTIAによる「ソフトウェア部品表の共通フォーマット策定に関する文書」や、MITREの医療機器SBOM運用ホワイトペーパーが参考資料として活用されています。
継続的な脆弱性監視体制
SBOMの提出義務化により、市販後も最新SBOMを保守して新たな脆弱性情報を監視する体制が必要です。専門の脆弱性データベース監視、ISAC(情報共有分析センター)からのアラート受信など、タイムリーな情報収集の仕組み構築が求められます。
市販後セキュリティ管理の強化
脆弱性モニタリングと協調的脆弱性開示
製品が市場に出た後も、メーカーは関連する脆弱性情報を継続的にモニタリングし、リスク評価を行う必要があります。外部の研究者やユーザから脆弱性報告を受け付けるCVD制度の整備、報告窓口の設置、社内対応手順の定義が推奨されます。
ソフトウェアアップデートとパッチ提供
デバイスの安全性に影響するサイバー脆弱性が発見された場合、適切な期間内にアップデートやパッチを提供することが求められます。特に患者へのリスクが高い重大な脆弱性は可能な限り迅速に緊急パッチをリリースし、そうでない脆弱性も定期的なアップデートサイクルで修正する計画が必要です。
製品の全ライフサイクル(TPLC)を通じたパッチ計画を策定し、通常時の定期更新と緊急時の臨時更新の双方を考慮します。アップデート提供手段としては、遠隔ソフトウェア更新など、リコールに至らないフィールドアップグレードが想定されます。
レガシーデバイスへの影響
法律上、新規制は将来の申請に適用されるため、既に市場に出ている承認済み機器は直接の義務対象ではありません。しかし、品質システム規則や一般安全性要求の下で、重大なサイバー脆弱性が判明した場合には適切に対処する責任が引き続き課せられています。
また、ソフトウェア変更等でFDAへの追加申請が必要となる場合には524B要件への適合が求められるため、事実上多くの既存製品も将来の変更時に準拠を迫られる可能性があります。
設計段階からのセキュリティ確保
脅威モデルの構築
開発初期にデバイスの使用シナリオや接続環境を分析し、想定されるサイバー脅威を洗い出します。各脅威に対するリスク評価を行い、リスクが高い部分に重点対策を割り当てることで、効率的なセキュリティ確保が可能になります。
セキュリティ設計要件の定義
脅威モデルに基づき、製品に必要なセキュリティコントロールを設計要件として明文化します。ユーザ認証の実装、患者データの暗号化、ソフトウェアのSecure Bootなど、具体的な対策を要件化します。ISO 81001-5-1(医療機器のサイバーセキュリティ規格)やNISTのサイバーセキュリティフレームワークなどの業界標準も参考にします。
実装とテストの徹底
セキュリティ要件に沿ったソフトウェア実装を行い、コード解析や侵入テストによる検証を重ねます。特にインターネットに直結する機器では、外部からのペネトレーションテストやFuzzテストによって脆弱な点を発見・修正することが重要です。
業界への影響と実務対応
社内体制の強化
多くの医療機器メーカーは社内に専門のセキュリティチームや役職(Product Security Officer等)を設置し始めています。製品開発部門と品質保証部門が連携し、設計段階からセキュリティレビューを行う仕組みづくりが求められます。
プロセス・規程類の更新
既存の設計開発手順書、リスクマネジメント手順書、脆弱性対応手順などを見直し、FDAガイダンスに沿った内容にアップデートする必要があります。リスクマネジメントプロセスにサイバーセキュリティの評価項目を追加し、ソフトウェア変更時のサイバーリスク再評価手順を定めるなどの対応が必要です。
開発スケジュールへの影響
セキュリティ対策と追加文書作成に時間を要するため、新製品の開発期間が延びる可能性があります。特に中小のデバイスメーカーにとってはリソース負担が重く、外部のセキュリティコンサルタントや試験機関を活用するケースも見られます。
FDAは510(k)電子申請の技術スクリーニングでサイバーセキュリティ項目不備をチェックする運用に入っており、不備があれば開発・申請のリスケジュールを余儀なくされる可能性もあります。早い段階から規制要件を織り込んだ開発計画を立てることが重要です。
まとめ:継続的な責務としてのサイバーセキュリティ
2023年以降、米国FDAによる医療機器サイバーセキュリティ規制は、ガイダンスから実効性ある法的要求へと大きく転換しました。Section 524Bの施行によりSBOM提供や脆弱性対応計画が法的義務となり、最新ガイダンスで製品ライフサイクル全般にわたるセキュリティ対策実装が詳細に指南されています。
これらの措置は、増大するサイバー脅威から患者を守るために不可欠であり、産業界も概ね支持を表明しています。もっとも実務上は、メーカーに新たな負担と高度な専門知識が要求されるため、組織体制の強化やツール導入による効率化が課題となります。
重要なのは、サイバーセキュリティ確保が単なる規制順守チェックリストではなく、患者安全と製品有効性を維持するための継続的な責務である点です。FDAは今後も状況に応じてガイダンス改訂や追加規則策定を行う可能性があり、メーカーは最新情報を注視する必要があります。
各社は今回強化された要件を踏まえ、自社製品の設計・開発から市販後サポートに至るまでセキュリティを組み込んだプロセスへと移行することが求められます。それにより結果的に医療機器の信頼性と安全性が高まり、デジタルヘルス時代における患者ケアの質の向上につながると期待されます。
コメント